入力ファイルエディター(LAMMPS固有の設定)¶
Force-Field(力場)¶
入力ファイルエディターの Force-Field で力場の設定を行います。画面右下のメニュー  から Force-Field をクリックしてください。
から Force-Field をクリックしてください。
Type of Force Field で使用したい力場の種類を選ぶと、それに対応した設定項目が有効になりますので、設定を行ってください。
力場の設定後、設定内容に応じて各原子のパラメーターを調整する必要がある場合には、 Resolve Force Field の Apply ボタンが赤色になりますので、クリックして設定を反映させてください。
原子に電荷を設定しない力場の場合、 Request Charge をyesに設定することで、外部電場用の電荷を設定することができます。
Lennard-Jones¶
Non-Bonded Interactions 欄でLennard-Jonesポテンシャル ![E=4\epsilon [(\sigma /r)^{12} -(\sigma /r)^6]](../_images/math/db8ab27b7d1179d1b97668dbff58ae498ad6accf.svg) のパラメーター
のパラメーター 、
、 を設定してください。
を設定してください。
Charge & Lennard-Jones¶
Lennard-Jonesポテンシャルのパラメーターに加え、電荷を設定して下さい。全電荷が0でない場合、 Resolve Force Field の Apply ボタンをクリックすると、全電荷が0になるように一定の値を差し引きます。
OPLS-AA¶
Resolve Force Field の Apply ボタンをクリックすると、OPLS-AAパラメーターセットに基づき、電荷およびLennard-Jonesポテンシャルのパラメーターを設定します。その後、全電荷が0になるように一定の値を差し引きます。
Non-Bonded Interactions 欄の各行を右クリックすると、割り当ての詳細を確認できます。
NeuralMD¶
Atomic Energy を without bias に設定することで、LAMMPS実行時にニューラルネットワーク最終層のバイアス項(原子の内部エネルギーに相当する定数)を0にし、原子エネルギーの平準化を図る機能が有効になります。
Potential File で設定する力場ファイルはAdvance/NeuralMDで生成するか、学習済みの力場ファイルを力場データベースからダウンロードして使うこともできます。
ヒント
NeuralMD力場は、GPUを使って計算を高速化することができます。
(Linuxのみ)ローカルで実行する場合、 の Number of GPU に使用するGPUの数を設定します。複数のGPUを使用する設定の場合、MPI並列のプロセスを各GPUに均等に割り当てて実行されます。0を設定するとGPUを使用しません。
リモートで実行する場合、SSHサーバーの設定で使用するキューのGPU設定を有効にしてください。
注釈
GPUドライバを事前にインストールしておく必要があります。CUDA 11.4.4を使用しており、これに対応するドライババージョン470.82.01以上が必要です。
元素数が5以上の場合は、力場作成時に重み付き対称関数を使っている必要があります。
ここでの設定はNeuralMD関連の計算にのみ適用されます。それ以外のグラフニューラルネットワーク力場のGPU使用の設定とは独立しています。これらのGPU使用についてはそれぞれの環境設定方法を参照してください。
汎用グラフニューラルネットワーク力場¶
オープンソースで公開されている、グラフニューラルネットワークに基づく学習済みの汎用力場を使います。
MatGL
M3GNet
CHGNet
MACE
MACE-Osaka24
MACE-OFF
Orb
MatterSim
FAIR-Chem
EquiformerV2
LAMMPSからPythonを呼び出して実行するため、事前にPython環境の設定が必要である他、いくつか制約があります。  をクリックすると説明が表示されます。
をクリックすると説明が表示されます。
MPI並列には非対応です(OpenMP並列・GPUを使った計算が可能です)。
一部のモデルでは、ビリアル応力が計算できないため、NPT・NPHアンサンブルでの計算、およびセルの最適化には非対応です。
Model には同梱されている学習済みモデルが表示されます。また、画面左上のアイコン  から でモデルをインポートできます。
から でモデルをインポートできます。
SevenNet¶
SevenNet として公開されている、グラフニューラルネットワークに基づく学習済みの汎用力場を使います。
マルチGPUに対応しており、1GPUあたり1MPIプロセスでの実行が想定されています。
SevenNetに対応したLAMMPSの実行ファイルはNanoLaboに同梱されておりません。リポジトリの説明に従ってリモートサーバー上に実行環境を用意し、実行ファイルを lammps_sevennet というファイル名で用意してください。必要に応じ、Job Script設定画面で PATH を設定してください。
Model にはSevenNetに同梱されている学習済みモデルが表示されます。また、画面左上のアイコン から でモデルをインポートできます。
Scheme(計算過程)¶
入力ファイルエディターの Scheme で計算過程の設定を行います。画面右下のメニュー から Scheme をクリックしてください。
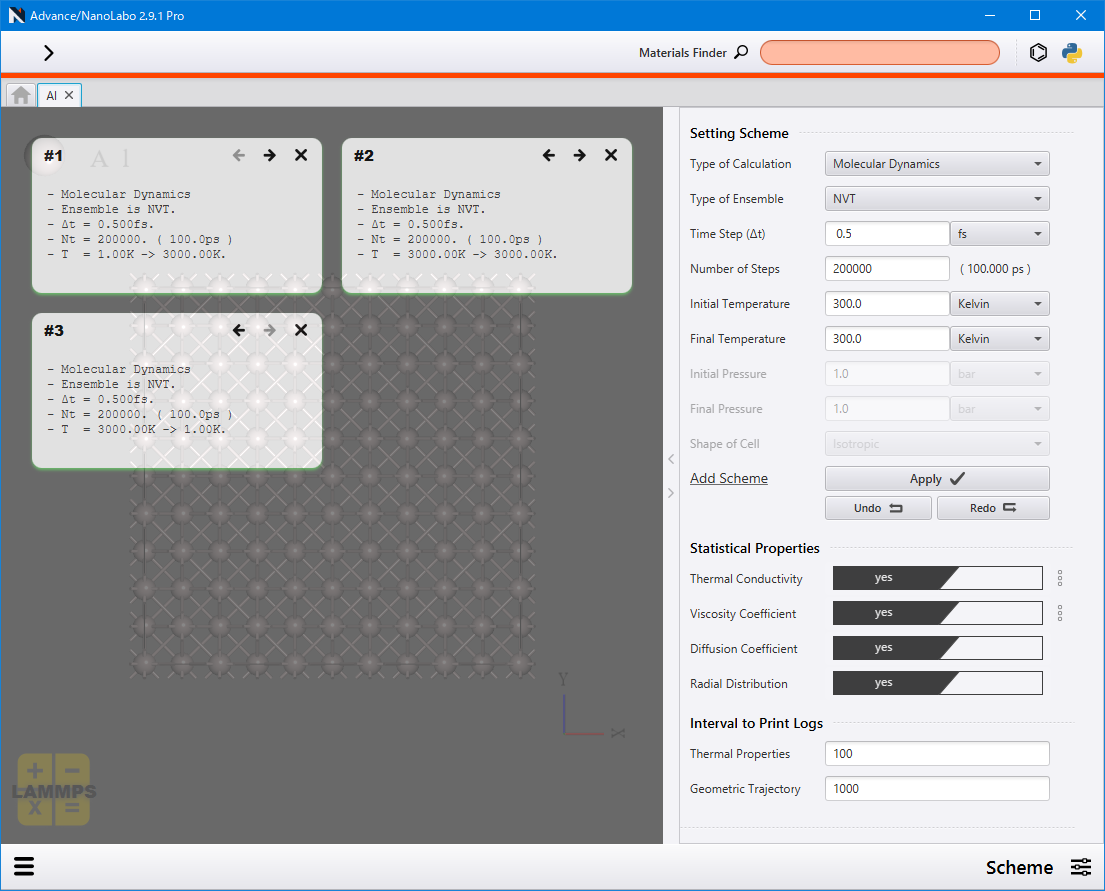
画面右側の Setting Scheme で計算過程のパラメーターを設定します。
Shape of Cell: NPH、NPTにおけるセル変形の拘束条件の設定
Isotropic
等方的な変形を許す
Anisotropic
x,y,z軸方向の伸縮を許す
Triclinic
任意の変形を許す
2つの軸方向の同じ割合での伸縮を許す
2つの軸方向の伸縮を許す
2つの軸を含む面内の任意の変形を許す
1つの軸方向の伸縮を許す
Add Scheme の Apply をクリックすると、画面左側に設定内容がタイル状に表示されます。必要に応じ、設定 → Apply を繰り返して過程を追加していきます。
タイルの左上の数字が過程の実行される順番です。前へボタン  、後へボタン
、後へボタン  で順番を入れ替えます。削除ボタン
で順番を入れ替えます。削除ボタン  で削除します。
で削除します。
また、ショートカットキーによる操作ができます。
操作 |
|
|---|---|
直前に追加したタイルを削除 |
Ctrl + C , Backspace , Delete |
アンドゥ(直前の操作の取り消し) |
Ctrl + Z |
リドゥ(取り消した操作のやり直し) |
Ctrl + Shift + Z |
* macOSでは Ctrl → command と読み替えてください。
追加済みのタイルの設定を後から編集するには、タイルをダブルクリックしてください。 Set the scheme ウィンドウが表示され、設定を編集できます。
また、 Statistical Properties の各項目を yes に設定することで、統計量(熱伝導率、粘性係数、拡散係数、動径分布関数(RDF))が計算され、結果画面に表示されます。熱伝導率、粘性係数については、  からパラメーターを設定できます。
からパラメーターを設定できます。
各熱力学量や原子構造は毎ステップではなく、間を空けて出力されます。 Interval to Print Logs で出力の間隔をステップ数で設定できます。
Option(追加操作)¶
入力ファイルエディターの Option で外場・外力等の設定を行います。
- E-Field
静電場を印加します。
- Ext. Forces
外力を印加します。
- Move Atoms
原子を一定の速度で(原子に働く力に依らず)動かします。
- Deform Cell
セルを一定の速度で変形させます。対象とする原子はセルの変形に追随します。
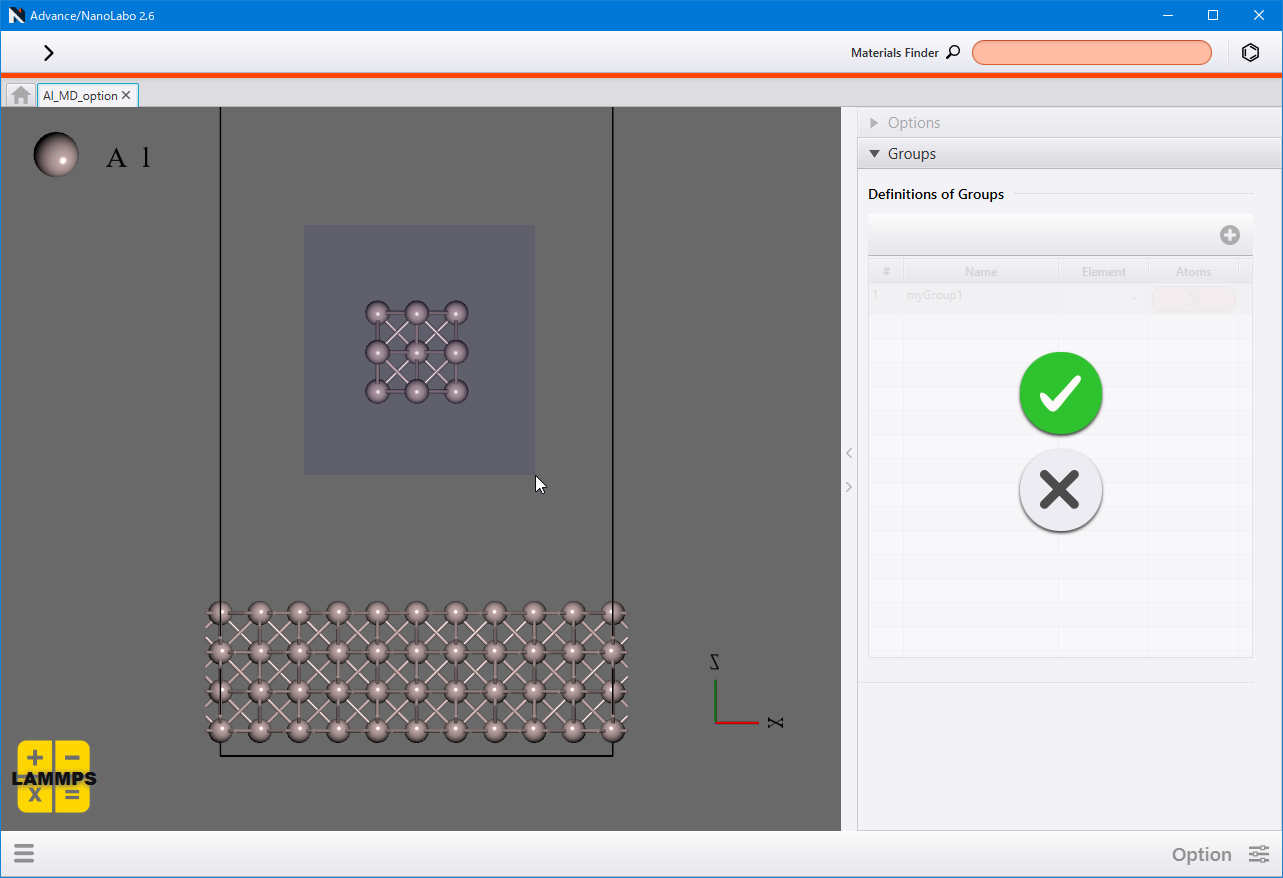
各タブの Target Group で対象とする原子グループを選び、パラメーターを入力します。
これらの設定は、 Scheme の過程ごとに独立しており、 Target Scheme で選択されている過程にのみ適用されます。
ヒント
初速度を与えて原子を動かすにはこの画面ではなく、 Geometry の Velocities で設定します。
また、 Target Group として最初はall(全ての原子)のみが選択できますが、Groups で原子グループを定義することができます。  をクリックして行を追加し、 Element で元素を選択して一括で指定するか、 Atoms のボタンをクリックして原子を選択して指定します。原子グループを削除するには行の右クリックメニューから Delete をクリックします。
をクリックして行を追加し、 Element で元素を選択して一括で指定するか、 Atoms のボタンをクリックして原子を選択して指定します。原子グループを削除するには行の右クリックメニューから Delete をクリックします。
注釈
LAMMPSの仕様により、原子グループ数の上限は32個(allを除いて、31個まで追加可能)となります。
User's(ユーザー定義)¶
入力ファイルエディターの User's で任意コマンド追加、任意変数出力・グラフ化の設定を行います。
- User's Additional Settings into Input-file
テキストボックスに任意のLAMMPSコマンドを追加すると、入力ファイルに挿入されます。
- User's Variables to Output and Plot
変数を追加すると、結果画面にボタンが追加され、時系列プロットとして表示することができます。また、CSVファイルにもカラムが追加されます。
変数としてはあらかじめ定義されているもの(LAMMPSマニュアル参照)や、LAMMPSコマンドで追加したものを使えます。
例として、bond・angleが定義されている計算(OPLS-AA力場)で、各時刻の結合距離・結合角の平均をプロットし、ヒストグラムをファイル出力する設定を示します。
User's Additional Settings into Input-file
compute 1 all bond/local dist compute 2 all angle/local theta compute 3 all reduce ave c_1 c_2 fix 1 all ave/histo 10 10 100 0.8 1.1 20 c_1 mode vector file dist.histo fix 2 all ave/histo 10 10 100 80 120 20 c_2 mode vector file theta.histo
User's Variables to Output and Plot
c_3[*]
LAMMPSコマンドの使い方についてはLAMMPSのマニュアルを参照してください。