入力ファイルエディター(Quantum ESPRESSO固有の設定)¶
擬ポテンシャルの設定¶
画面右下のメニュー  から Geometry を選び、 Elements の Element's Properties で擬ポテンシャルの設定を行います。各元素の Pseudopotential 欄にあるボタンをクリックすると、擬ポテンシャルファイルを選択できます。また、設定に応じたカットオフエネルギーの推奨値が下部に表示されます。
から Geometry を選び、 Elements の Element's Properties で擬ポテンシャルの設定を行います。各元素の Pseudopotential 欄にあるボタンをクリックすると、擬ポテンシャルファイルを選択できます。また、設定に応じたカットオフエネルギーの推奨値が下部に表示されます。
擬ポテンシャルファイルを追加する場合は、左上メニュー  から でインポートします。
から でインポートします。
擬ポテンシャル変更後、 SCF の設定で Cutoff 入力欄のデフォルト値設定ボタン  をクリックすると、更新されたカットオフエネルギーの推奨値が入力されます。
をクリックすると、更新されたカットオフエネルギーの推奨値が入力されます。
バンド計算の経路¶
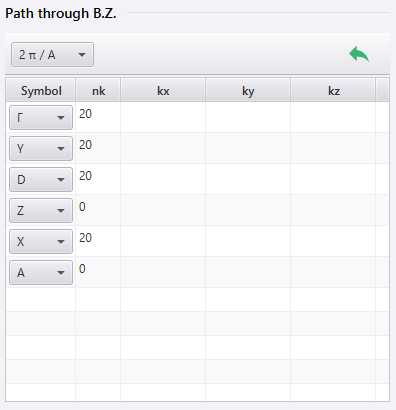
画面右下のメニュー から Band または Ph. Dispersion を選ぶと、設定項目の中に Path through B.Z. (バンド計算の経路)があります。この設定方法を説明します。
Symbol では、経由する点を指定します。リストボックス中に無い任意の点を指定したい場合は、 Symbol を空白にし、座標を kx, ky, kz (または qx, qy, qz )に入力します。
点の追加、削除、入れ替えは行の右クリックメニューから行います。追加の際は、クリックした行の次に新しい行が挿入されます。
nk (または nq )には、その行と次の行で指定した点の間で計算する点の数を指定します。例えば画像の1行目では nk に20と入力していますが、これはその行で指定しているΓ点と、次の行で指定しているY点の間の20点で計算することを表します。
また nk に0を入力すると、その行と次の行で指定した点の間では計算を行いません。バンド図上では、その行と次の行の点が重なって表示されます。
ESM法の設定¶
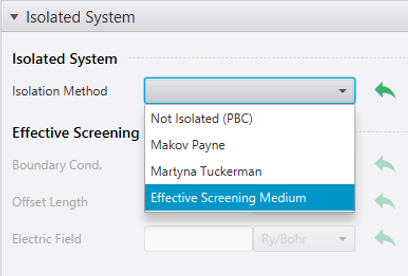
画面右下のメニュー から SCF を選ぶと、設定項目の中にIsolated Systemがあります。Isolated Systemの中にあるIsolation MethodからEffective Screening Method(ESM法)を選びます。
ESM法を有効にすると、計算の都合上、原子構造がスラブ面の法線方向にシフトすることがあります。
NEB法の設定¶
画面右下のメニュー から NEB を選ぶと、設定項目の中にImagesがあります。ImagesではNEB法の計算に用いる初期イメージの編集を行います。この設定方法を説明します。
はじめは現在のプロジェクトの構造のみがイメージ(First)に追加されています。この状態で  をクリックするとプロジェクトの選択画面が出るので、他端のイメージ(Last)を指定します。このとき中間イメージ(Intermediate)が自動で生成されます。
をクリックするとプロジェクトの選択画面が出るので、他端のイメージ(Last)を指定します。このとき中間イメージ(Intermediate)が自動で生成されます。
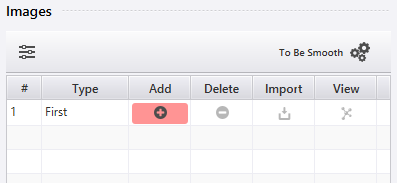
個別のイメージに対する操作は各行のボタンから行います。
イメージを追加するには
をクリックします。選択中のイメージの後に新規イメージが挿入されます。イメージを削除するには
 をクリックします。
をクリックします。イメージを別の構造に置き換えるには、
 をクリックして、置き換えたい構造を含むプロジェクトを選びます。
をクリックして、置き換えたい構造を含むプロジェクトを選びます。イメージの構造を確認するには、
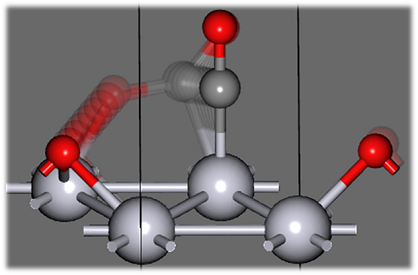
 をクリックします。選択中のイメージ以外は残像で表示されます。
をクリックします。選択中のイメージ以外は残像で表示されます。
イメージ全体をスムーズ化するには、  をクリックします。
をクリックします。
ヒント
正しくNEB計算を行うには、各イメージの対応する原子が入力ファイル内で同じ順序に並んでいる必要があります。イメージの追加時には原子間の結合を元にして自動的に原子の並び替えを行います。このアルゴリズムは  ボタンから調整できます。
ボタンから調整できます。
Bond Watching: イメージ追加時に原子の並び替えを行うかどうかの設定です。 no に設定すると、元の入力ファイルの原子の順序がそのまま使われます。
Bond Scale: 原子間の距離から結合の有無を判定するときの係数です。
XAFSの設定¶
画面右下のメニュー から XAFS を選ぶと、XAFSの設定画面が表示されます。ここではXAFS特有の設定について説明します。
Super Cell は、励起される原子どうしの距離を十分にとるための設定です。セルが小さい場合、周期境界条件により隣り合うセル中の励起原子からの影響を受けてしまいますが、自動的にスーパーセルを作ることでこれを防ぎます。スーパーセルの繰り返し回数はセルサイズに基づいて適切な値が自動的に入ります。
Target Atom で励起される原子を選択します。 Select Atom ボタンをクリックし、原子をダブルクリックして選択します。 End Selecting Atom をクリックすると、選択した原子がボタンに表示されます。
次に、 Core-hole P.P. のボタンをクリックし、励起される原子に対する擬ポテンシャルを選択します。ここでは励起された後の、内殻にホールがある擬ポテンシャル(コアホール擬ポテンシャル)を選択する必要があります。NanoLabo Toolにはコアホール擬ポテンシャルは同梱されていませんので、必要に応じ Download ボタンからダウンロードしてください。通常、ファイル名に「star + 軌道名」(例えばstar1s)のようにホールの位置(=励起元の軌道)が入っています。
また、XAFSの計算には内殻の波動関数の情報を使うため、その情報を含むGIPAW擬ポテンシャルを設定する必要があります。 画面右下のメニュー から を開き、励起される原子の元素にGIPAW擬ポテンシャルを設定してください。通常、ファイル名に「gipaw」が入っています。
CPMDの設定¶
画面右下のメニュー から MD を選び、 CPMD タブの Car-Parrinello をyesに設定することで、Car-Parrinello法を使った分子動力学計算を行います。
最初に電子状態のSCF計算を行い、そこから電子状態と原子構造を時間発展させます。最初のSCF計算の手法は Initial SCF で設定します。その他、電子状態計算のパラメーターは SCF 画面での設定内容を参照して決められますが、CPMDの制約に合わせて変更されることがあります。
また、擬ポテンシャルについて以下の制約があります。
ウルトラソフト擬ポテンシャルを使う場合は、box gridを設定する必要があります。
PAW擬ポテンシャルは使えません。
NMRスペクトルの設定¶
画面右下のメニュー から GIPAW を選ぶと、NMRスペクトルの設定画面が表示されます。
Macroscopic Shape をyesにすると、試料のマクロな形状を考慮した補正を行います。 Shape Tensor で形状に応じた各軸方向の補正を指定します。
参照物質の遮蔽テンソル  は、結果プロット画面で設定できます。
は、結果プロット画面で設定できます。
フォノン分散の設定¶
画面右下のメニュー から Ph. Dispersion を選ぶと、フォノン分散の設定画面が表示されます。
DOS タブの Calc. DOS 、 BAND タブの Calc. Band をそれぞれyesに設定することで、フォノン状態密度、フォノンバンドの計算を行います。
Non-Analytic Term をyesに設定すると、LO-TO分裂を考慮した計算を行います。補正を有効にするために、あらかじめ Phonon でΓ点 (qx, qy, qz) = (0, 0, 0) 、Dielectric Constant ・ Effective Charge をyesに設定して計算を行い、ボルン有効電荷を得ておく必要があります。ボルン有効電荷が計算されていない場合、 Non-Analytic Term がyesでも補正なしの計算になります。